La Enfermedad de Fabry (EF) es una enfermedad rara lisosomal causada por mutaciones en el gen GLA, ubicado en el cromosoma X. Estas mutaciones resultan en una reducción de la actividad de la enzima alfa-galactosidasa A.
Como consecuencia, se origina una patología progresiva y multisistémica caracterizada por manifestaciones renales, cardiovasculares, cutáneas y cerebrovasculares en diferente grado.
Angioqueratomas, una manifestación cutánea común en la enfermedad de Fabry.
¿Qué es la Enfermedad de Fabry?
La enfermedad de Fabry (EF) es una enfermedad genética lisosomal que se caracteriza por la acumulación de globotriaosilceramida en una gran variedad de células.
Dentro de la célula, estos lípidos (concretamente unos llamados glicoesfingolípidos), se acumulan en los lisosomas, que es el lugar de la célula donde son degradados normalmente.
La enfermedad de Fabry es una enfermedad de depósito lisosomal de carácter hereditario, ligada al cromosoma X, causado por el déficit de la enzima alfa-galactosidasa A (alfa-GLA A), lo que conduce a la acumulación de glicoesfingolípidos, principalmente globotriaosilceramida, en los tejidos.
La EF es de difícil diagnóstico debido a la heterogeneidad de sus variaciones clínicas, donde predominan las manifestaciones clásicas como: dolor digital, acroparestesias, manifestaciones cutáneas que incluyen angioqueratomas e hipohidrosis, trastornos vestibulares y cocleares, proteinuria, enfermedad renal crónica que requiere terapia sustitutiva, hipertrofia ventricular izquierda, arritmias y enfermedad cerebrovascular que a menudo son descartados como simulación o son atribuidos erróneamente a otros trastornos.
Es una enfermedad multisistémica (riñón, corazón, sistema nervioso, piel, ojos, entre otros) que requiere atención multidisciplinaria. Varios especialistas deben trabajar conjuntamente.
Los síntomas aparecen de forma progresiva. Los profesionales deben considerar esta posibilidad, detectarla y diagnosticarla a tiempo.
Herencia y Afectación
La enfermedad de Fabry es genética, crónica y progresiva y tremendamente nociva en la vida familiar de nuestros pacientes.
La herencia es peculiar porque se hereda ligada al cromosoma X, de tal forma que las madres, además de padecer la enfermedad, la transmiten.
Ya que las mujeres tienen normalmente dos copias del gen X, y los hombres solo una, la enfermedad suele afectar más a hombres que a mujeres.
Las mujeres, al tener dos cromosomas X, pueden tener que el cromosoma no afectado compense parcialmente al alterado. Por ello, pueden presentar diversas formas clínicas, de mayor o menor gravedad, y generalmente con aparición más tardía que en los hombres.
Cuando el padre (XY) está afectado, transmite su único cromosoma X a sus hijas, pero no a sus hijos, que reciben el cromosoma Y del padre. Cuando la madre (XX) está afectada, al tener dos cromosomas X y solo uno alterado, tiene un 50% de probabilidad de transmitir tanto el cromosoma X sano como el alterado a hijas e hijos.
La herencia ligada al cromosoma X hace que haya casi el doble de mujeres afectadas que hombres, pero la variabilidad en la expresión provoca que muchas mujeres no sean diagnosticadas.
Los hombres con mutaciones clásicas de la enfermedad de Fabry suelen presentar formas graves, con inicio incluso en la infancia o adolescencia.
Prevalencia y Diagnóstico
Se estima que la incidencia real de esta patología está infradiagnosticada, sobre todo en mujeres.
La enfermedad de Fabry es una enfermedad considerada rara o minoritaria. Tiene una prevalencia en torno a 1/100.000 o 125.000 recién nacidos.
Debido a su baja incidencia y a la variedad de sus síntomas, los expertos consideran que afecta a muchas más personas de las que se diagnostican.
El principal nicho de nuevos diagnósticos es el estudio de familiares de pacientes afectos.
Es obligado hacer un árbol genealógico completo para conocer y diagnosticar de una forma precoz a los familiares de pacientes con enfermedad de Fabry porque es la única manera que tenemos de anticiparnos al desarrollo de lesiones en órganos diana.
El diagnóstico precoz puede realizarse mediante análisis de sangre para determinar la actividad enzimática y estudios genéticos.
La confirmación de una mutación en el gen GLA puede realizarse por medio de estudios genéticos, aunque por ahora, no es una práctica generalizada.
Los estudios genéticos ampliados ayudan a analizar la familia y detectar individuos afectados en fases tempranas. Es fundamental el cribado de poblaciones de riesgo, como personas con enfermedad renal de origen desconocido, hipertrofia miocárdica inexplicada, ictus precoz o pérdida auditiva súbita.
Se conocen aproximadamente 1.000 variantes diferentes del gen GLA afectado. Esto explica por qué la enfermedad de Fabry puede presentarse en formas diversas según el tipo de mutación (formas clásicas o formas tardías/atípicas).
Manifestaciones Clínicas
Siendo una enfermedad multisistémica, las manifestaciones clínicas son variadas, pudiendo aparecer en la infancia o adolescencia (cutáneas, cardiacas, renales, neurológicas, etc.) con un orden de aparición variable.
Las mutaciones clásicas comienzan en la infancia, y los niños pueden presentar síntomas desde los 3-4 años, según el tipo de mutación.
En los niños las manifestaciones de la enfermedad de Fabry fundamentalmente son en forma de dolor neuropático. Se conoce como crisis de Fabry y es dolor urente o quemante en palmas de las manos y plantas de los pies, junto con a veces fiebre o fallo de medro.
Además, de niños esta enfermedad no afecta de forma evidente a los órganos diana, aunque sí les está afectando desde el punto de vista histológico. En la edad adulta es cuando aparece el daño en órganos vitales como el corazón y el riñón.
En esta enfermedad es cierto que no hay una alteración del nivel cognitivo y nuestros pacientes son igual de inteligentes que si no la tuviesen, lo que ayuda bastante a la hora de entender y de colaborar en la enfermedad.
Las manifestaciones de inicio predominantes en nuestra serie son el angioqueratoma, hipertrofia ventricular izquierda, HTA y la enfermedad renal. Las manifestaciones vasculares que predominan en el seguimiento son la HTA, hipertrofia ventricular izquierda, la arteriopatía periférica y las manifestaciones cutáneas, oftalmológicas y digestivas.
Síntomas Comunes
- Afectación del sistema nervioso periférico: Dolor y molestias en manos y pies. Con calor o al caminar, pueden enrojecerse y doler (acroparestesias).
- Pequeñas manchas rojas elevadas (angioqueratomas): No contagiosas, generalmente en abdomen, genitales, muslos y glúteos.
- Sudoración reducida (hipohidrosis): Provoca baja tolerancia al calor.
- Rubor (flushing): Debido a alteración de la regulación de la temperatura.
- Intolerancia al ejercicio: Los pacientes se cansan muy rápidamente.
- Manifestaciones gastrointestinales: Dolor abdominal desde la infancia, a veces con diarrea relacionada con la alimentación; las comidas grasas empeoran los síntomas. Similares al síndrome de intestino irritable.
- Afectación cardíaca: Hipertrofia miocárdica (corazón agrandado) sin causa aparente; arritmias (alteraciones del ritmo cardíaco).
- Afectación renal: Disminución de la función renal, pérdida de proteínas por la orina.
- Afectación del sistema nervioso central: Accidente cerebrovascular. Si una persona relativamente joven tiene un ictus sin causa clara, se debe considerar la enfermedad de Fabry.
- Pérdida auditiva: Súbita o rápidamente progresiva.

Infografía que muestra los diversos síntomas de la Enfermedad de Fabry.
Manifestaciones cutáneas
La EF fue descrita por primera vez en 1898 por los dermatólogos Johan Fabry y William Anderson, reportando lesiones cutáneas generalizadas a las que denominaron angioqueratomas corporales difusos.
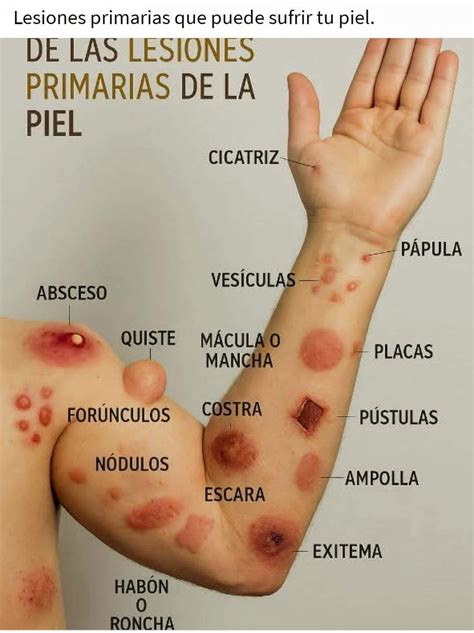
- Lesiones cutáneas o angioqueratomas: son puntos rojos situados entre el ombligo y las rodillas. Aparecen durante la adolescencia y pueden aumentar con la edad.
- Telangiectasias: Se aprecian telangiectasias en el rostro, los labios y la mucosa oral.
En el examen físico se evidencian pápulas rojo-violáceas generalizadas, algunas de ellas agrupadas en el rostro y el borde bermellón.
Tratamiento
No existe una cura completa de la enfermedad. El tratamiento actual consiste en proveer al paciente de la enzima alfa-Gal.
Aparte del tratamiento enzimático sustitutivo, es necesario tratar cada síntoma en particular como el dolor, la insuficiencia renal o problemas cardíacos.
La terapia médica no específica y quirúrgica es eficaz para ralentizar el deterioro o compensar la insuficiencia orgánica en los pacientes con enfermedad de Fabry.
En el mejor de los casos, los pacientes que conviven con esta enfermedad pueden no necesitar tratamiento específico, pero van a tener un seguimiento anual. En el peor, algunos pacientes pueden requerir un trasplante renal.
En ausencia de tratamiento, los órganos vitales como los riñones, el corazón y el cerebro con el tiempo comienzan a deteriorarse y pueden surgir complicaciones graves.
Desde 2001, está disponible la terapia de reemplazo enzimático (ERT). Esta condición provoca una producción deficitaria de la enzima. Mediante ingeniería genética, la enzima se produce en laboratorio y se administra por vía intravenosa cada 15 días. La proteína entra en la célula y actúa durante varios días, pero dado que el organismo no la produce, es necesario seguir administrándola por vía intravenosa.
La ERT reduce la aparición de complicaciones graves y mejora la supervivencia en una media de 20 años. Para obtener un beneficio óptimo, el tratamiento debe iniciarse antes de que se produzcan daños irreversibles en los tejidos.
Recientemente, ha surgido otra alternativa: la terapia con chaperonas farmacológicas. Si la mutación del gen no produce proteína, este tratamiento no puede utilizarse. Si se produce una proteína defectuosa, la molécula se une a ella, modificando su estructura y mejorando su actividad en variantes genéticas respondedoras (20-30%).
Otro tratamiento en desarrollo es la terapia génica, que consiste en introducir un gen normal en la célula mediante un vector para que la célula produzca la enzima de forma normal y permanente. Si tiene éxito, sería un tratamiento curativo.
En el año 2001 empezó el tratamiento enzimático sustitutivo, ha supuesto un antes y un después en el curso de la enfermedad de Fabry. La esperanza de vida de los pacientes varones, que suelen tener una enfermedad más grave, antes era de 15 años menos que la que tienen ahora y en las mujeres de 10 años menos.
Después, apareció la terapia con chaperonas y ahora se está investigando la terapia de reducción de sustrato y la terapia génica, donde hay algunos ensayos en fase I y fase II.
Quiero recalcar la labor de nuestros pacientes, que son tremendamente generosos en la investigación. Saben que son enfermedades muy poco frecuentes y que deben prestarse siempre como parte del avance de la ciencia.
Asesoramiento Genético
El Asesoramiento o Consejo Genético contribuye a aliviar la incertidumbre y el sentimiento de vulnerabilidad que puede surgir ante el diagnóstico de la enfermedad, así como a la adaptación ante la condición genética familiar.
De este modo, la incorporación del Asesoramiento Genético en el sistema asistencial puede producir una mejora de la calidad de vida y en la prevención clínica de los pacientes.
Las intervenciones de Asesoramiento Genético de manera estructurada y dirigida en un entorno clínico controlado en pacientes con EF aumentaba tanto el Control de decisión (p = 0,015), así como el Control cognitivo (p = 0,024). Esto conducía a una mayor toma de decisiones y participación en las acciones de salud preventivas; principalmente en cribado genético familiar (p < 0,001).
Se ha comprobado que el aumento de participación en el cribado genético familiar incrementa la identificación de portadores -especialmente mujeres- anticipándose, de esta manera, a la aparición clínica de daños orgánicos irreversibles.
La importancia de la detección precoz
La Dra. Mónica López, jefa de sección del Servicio de Medicina Interna del Hospital Ramón y Cajal recalca la importancia de realizar el estudio de familiares de pacientes con enfermedad de Fabry ya que “es la única manera para anticiparse al desarrollo de lesiones en órganos diana”.
Su pronóstico mejora con tratamiento pero identificarla pronto evita complicaciones.
La EF precisa un alto índice de sospecha ya que las formas de presentación, con excepción de las dermatológicas, son muy inespecíficas.
El diagnóstico precoz puede cambiar el curso natural de la enfermedad cuando no exista enfermedad de órgano establecida.
Enfermedad de Fabry: cómo y cuándo tratar

Logo del Día de las Enfermedades Raras.